Welcome to the Brain Vascular Malformation Consortium

Join one of our research studies
Participants make it possible for researchers to find new treatments, speed diagnosis and improve the lives of those affected by rare diseases.
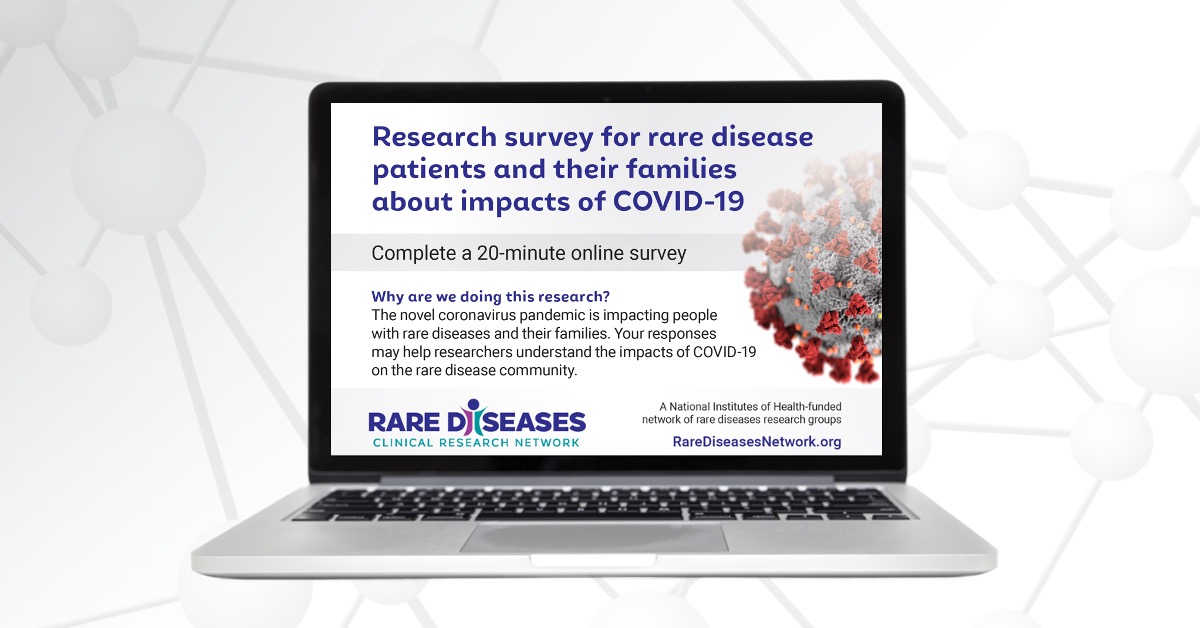
Find a StudyNational Survey Reveals Impact of COVID-19 on People Living with Rare Diseases and Their Families
The devastating impact of COVID-19 on the general population is well-documented—but less is known about the millions of people living with rare diseases.
Join the RDCRN for Rare Disease Day at NIH on February 29, 2024
Don’t miss the in-person and virtual celebration of Rare Disease Day at NIH on Thursday, February 29, 2024, from 9 am to 5 pm EST.

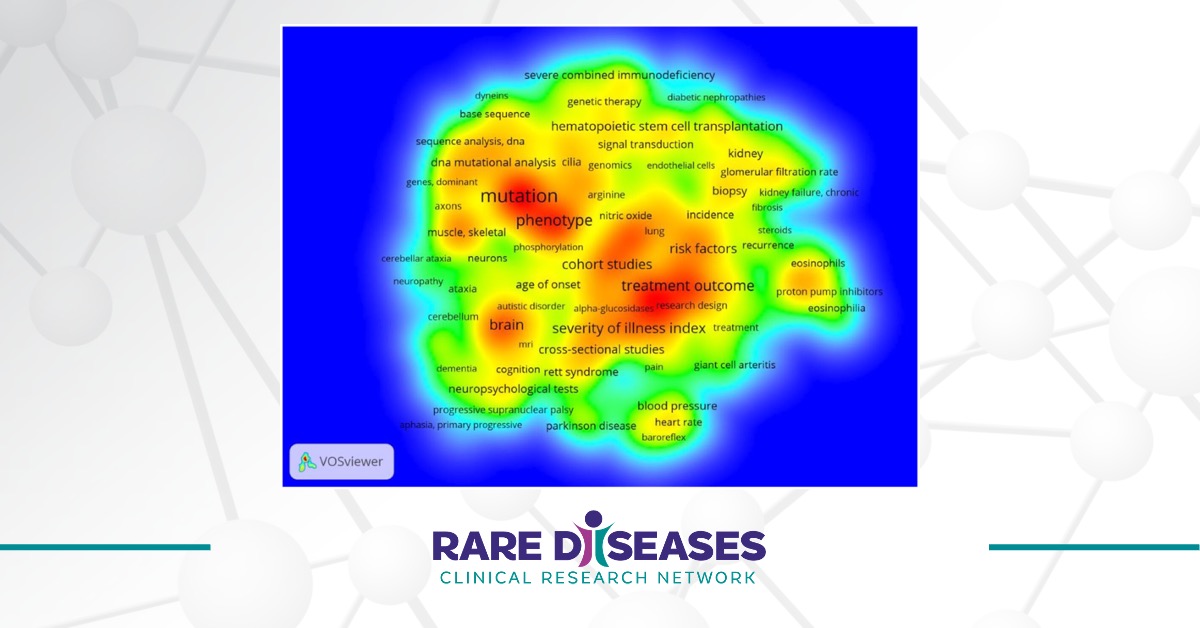
Twenty Years of the Rare Diseases Clinical Research Network: Looking Back, Looking Ahead
The Rare Diseases Act of 2002 (H.R. 4013) enacted the establishment of the Rare Diseases Clinical Research Network (RDCRN) in 2003.