
Expanding knowledge of the porphyrias by developing new strategies and methods for diagnosis, treatment, and prevention of illness and disability.

You Can Help
Discover opportunities to participate in research and help further innovations in care for porphyrias patients.
Find a StudyNational Survey Reveals Impact of COVID-19 on People Living with Rare Diseases and Their Families
The devastating impact of COVID-19 on the general population is well-documented—but less is known about the millions of people living with rare diseases.
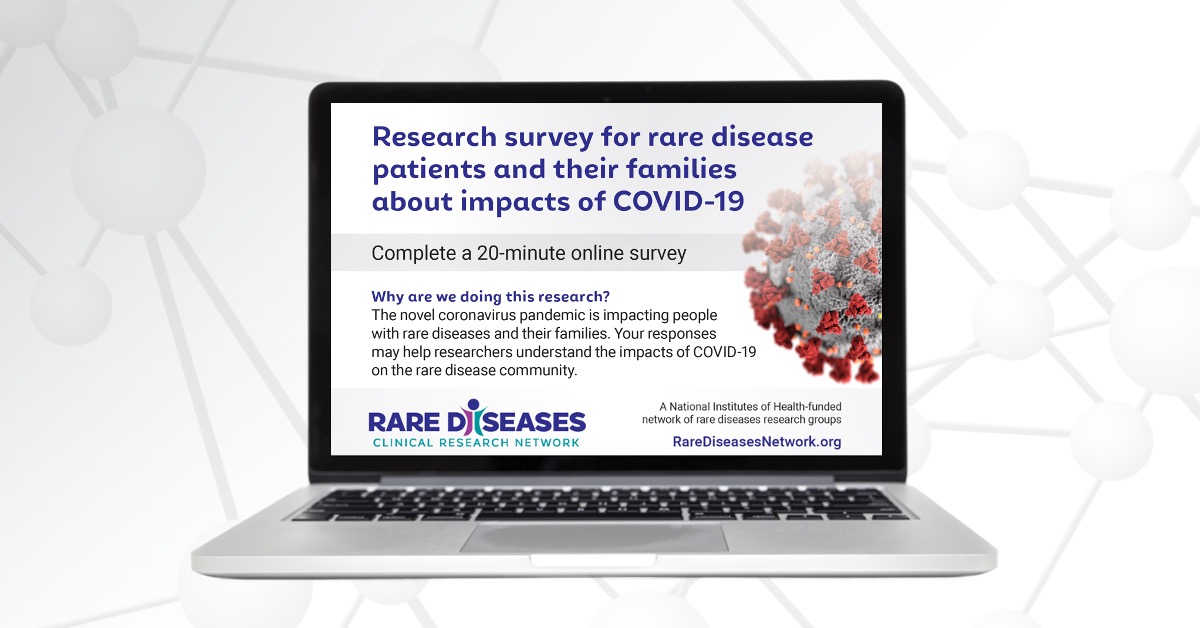
Join the RDCRN for Rare Disease Day at NIH on February 29, 2024
Don’t miss the in-person and virtual celebration of Rare Disease Day at NIH on Thursday, February 29, 2024, from 9 am to 5 pm EST.

Twenty Years of the Rare Diseases Clinical Research Network: Looking Back, Looking Ahead
The Rare Diseases Act of 2002 (H.R. 4013) enacted the establishment of the Rare Diseases Clinical Research Network (RDCRN) in 2003.
PC Pilot and Feasibility Program
The Porphyria Consortium provides a novel support mechanism for trainees, postdocs, and junior faculty who want to pursue new directions in porphyria. The PC leadership welcomes applicants from across the U.S. and will strive to provide a rapid review and constructive comments for applicants.