
Striving to improve the lives of individuals and families affected by urea cycle disorders.

Join One of Our Research Studies
Participants make it possible for researchers to find new treatments, speed diagnosis, and improve the lives of those affected by rare diseases
Find a StudyRecruiting Now
UCDC is currently recruiting for our Longitudinal Study of Urea Cycle Disorders.
Rare Research Report: April 2024
Each month, we share summaries of recent Rare Diseases Clinical Research Network (RDCRN) grant-funded publications.

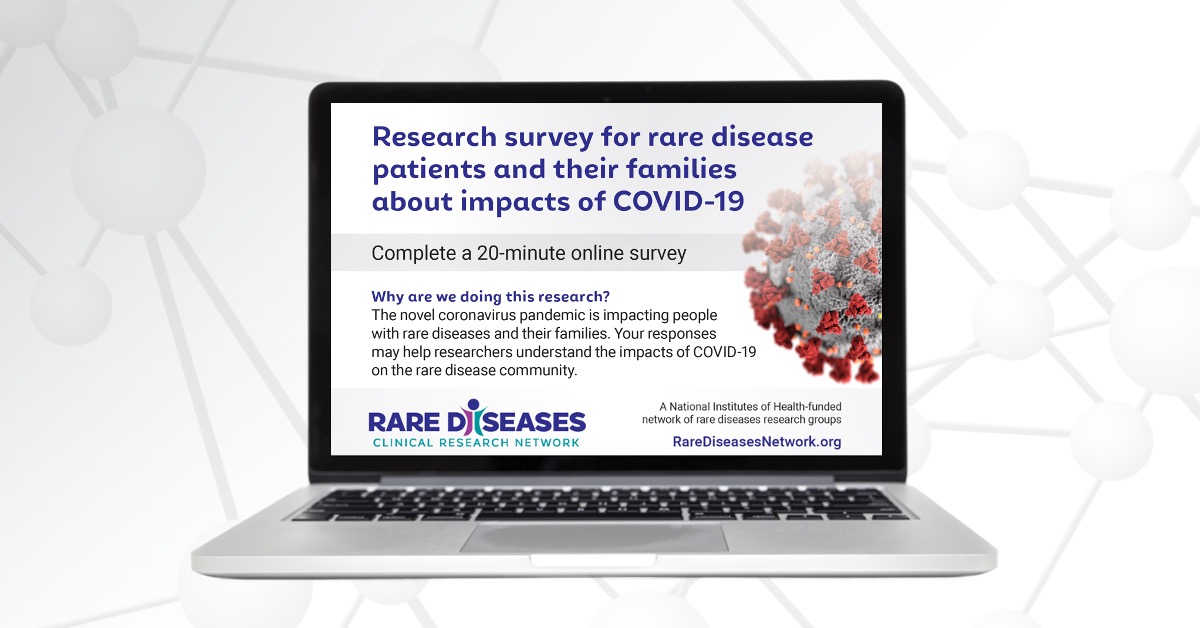
National Survey Reveals Impact of COVID-19 on People Living with Rare Diseases and Their Families
The devastating impact of COVID-19 on the general population is well-documented—but less is known about the millions of people living with rare diseases.
Join the RDCRN for Rare Disease Day at NIH on February 29, 2024
Don’t miss the in-person and virtual celebration of Rare Disease Day at NIH on Thursday, February 29, 2024, from 9 am to 5 pm EST.


Learn More About Urea Cycle Disorders
Access educational materials, frequently asked questions, lectures, and more.
Learn More